Quantitative Traits and QTL Mapping in Dogs
Most breeder-relevant traits do not come in clean on-or-off categories. They vary continuously across a population, which is why families rarely get a simple yes or no answer when they ask whether a dog is "good" on hips, temperament, or cancer risk. That continuous variation makes them quantitative traits, and it is why canine geneticists talk about QTL mapping, association studies, and polygenic risk models rather than clear carrier-affected-clear logic. Understanding this distinction is one of the single most useful conceptual shifts a buyer or breeder can make when reading modern canine genetic literature, because it separates the traits where single-mutation testing genuinely closes the question from the traits where even the best available testing only estimates probability. Documented
What It Means
Quantitative traits as a category
A quantitative trait is a trait that varies along a continuum rather than falling into a few discrete classes. Hip score, body size, trainability, fearfulness, coat color intensity, and many disease liabilities work this way. You cannot sort a population of Golden Retrievers into "has good hips" and "has bad hips" without imposing an artificial threshold somewhere in the middle of a continuous distribution, because the actual underlying biology produces a smooth bell-shaped range where most dogs cluster near the average and smaller numbers sit at each tail.
These traits are usually influenced by many loci of small or moderate effect, significant environmental inputs, interaction among loci (epistasis, where the effect of one gene depends on which alleles are present at another gene), and developmental history including prenatal and early-life conditions. That combined architecture is why quantitative genetics exists as a distinct branch of the field with its own mathematical tools rather than being handled by the same Punnett-square logic that works for simple Mendelian traits. The behavior of a trait shaped by fifty loci of small effect is qualitatively different from the behavior of a trait shaped by one locus of large effect, even when both are technically heritable. Documented
The distinction matters because the breeder's intuition developed on simple traits fails on complex ones. A breeder used to thinking about clear recessive conditions may assume that if they remove affected dogs from their breeding population, the trait will disappear quickly. For a single-locus recessive trait, that intuition is roughly correct. For a polygenic quantitative trait, it is not, because the contributing alleles are distributed across many dogs at moderate frequencies and no single culling decision dramatically shifts the population distribution. Progress on quantitative traits is measured in fractions of a standard deviation per generation rather than in binary elimination of affected individuals.
What a QTL actually is
A QTL, or quantitative trait locus, is a genomic region associated with variation in a quantitative trait. The wording matters. A QTL is not automatically a single causal gene. It is a region statistically associated with some amount of variation in the measured trait in the population that was studied, and the association may reflect a single causal variant, multiple variants in the same neighborhood, or a marker linked to the real cause somewhere nearby.
The size of the region varies depending on the mapping method, the sample size, the density of markers used, and the LD structure in the study population. Documented A fine-mapping study with dense markers and a large sample can narrow a QTL down to a relatively small region containing only a handful of candidate genes. A coarser association study may identify a QTL spanning hundreds of kilobases or more, containing many genes, without being able to pinpoint which one is carrying the signal. Both are QTL results in the technical sense, but they give breeders very different amounts of actionable information.
Researchers identify QTL through pedigree-based linkage mapping in families of known relationships, or through genome-wide association studies (GWAS) in populations of many unrelated individuals. Documented Each method has strengths. Linkage mapping works with smaller samples but requires pedigree structure. GWAS requires larger samples but does not need pedigree knowledge and can detect associations across an entire breed population at once. Modern canine studies often combine both approaches.
Why dogs are an attractive study system
Dogs are especially interesting for QTL work because breed structure makes some association signals easier to detect than they would be in more anciently outbred populations. Long linkage-disequilibrium blocks in breeds mean that fewer markers can sometimes capture larger trait-associated regions, and smaller sample sizes can reach statistical significance that would require much larger human cohorts to achieve. This is one of the reasons canine genetics has progressed so quickly over the last two decades relative to the number of dogs actually genotyped, and it is why dogs are sometimes used as a model system for traits researchers ultimately care about in humans.
That is the upside. The downside is that long linked blocks can make the signal broad rather than surgical. The associated marker may not be the causal variant. It may simply sit in a region traveling with the causal variant in the breed under study, and moving to a different breed with a different LD structure may weaken or eliminate the association entirely. A QTL therefore tells you where to look, not always what the final answer is, and the work of moving from a QTL region to a specific causal mechanism often takes years of additional fine-mapping and functional validation.
This is also why QTL mapping work is most useful as a research tool and only sometimes translates directly into breeder-facing tests. Documented A QTL can be real, replicable, and scientifically important while still being too broad or too breed-specific to form the basis of a commercial test that gives individual dogs useful predictions. The gap between "identified in a study" and "clinically useful for an individual dog" is wide enough that many research-grade QTL findings never make the transition.
What QTL mapping has revealed about canine complex traits
The most important thing QTL mapping has revealed about traits like hip dysplasia, cancer susceptibility, and many behavioral tendencies is that these traits are not simple one-gene stories hiding behind complex surface variation. They are genuinely distributed across multiple genomic regions with modest effect sizes, interacting with environment, developmental history, and population structure. No single QTL accounts for most of the variance in any of these traits, and the combined explanatory power of all identified QTLs for a given complex trait is usually well below 100 percent of the known heritable component, which means either additional QTLs of very small effect remain undiscovered or the architecture includes epistatic and environmental components that simple additive models do not capture.
This does not make the work weak. It makes it honest. The field is revealing the complexity rather than hiding it, and the honest complexity is more useful to breeders than a false simplicity would be. A breeder who understands that hip dysplasia is polygenic will make better decisions than a breeder who is waiting for a single "hip gene" test to be discovered, because the first breeder is using the right mental model for the real biology while the second is waiting for something that will likely never arrive in the form they are imagining.
For breeders and families, the practical consequence is that most DNA tests for complex traits are not diagnostic in the way a single-mutation retinal test can be diagnostic. At best, they estimate inherited liability using many associated loci, weighted by their individual effect sizes, summed into a polygenic risk score. Documented That can be useful as an additional layer of information on top of phenotypic screening, pedigree analysis, and line knowledge. It is still a probability statement about a continuous trait, not a verdict about a binary condition.
From QTL to polygenic risk scores
The frontier of canine complex-trait genetics is polygenic risk scoring, which attempts to combine many QTL signals into a single risk estimate for an individual dog. Polygenic scores have become standard in human genetics over the last decade and are beginning to appear in canine contexts for traits like hip dysplasia and some cancer predispositions. When well constructed and properly validated, they can meaningfully improve risk estimation beyond pedigree information alone.
But polygenic scores also come with interpretation cautions that families and breeders should understand. They are probability estimates, not diagnoses. They are most accurate in populations similar to the one in which they were developed, which for dogs means breed-specific scores often work poorly across breeds. They capture the additive genetic component the study identified, which may be only part of the total heritable architecture. And they do not account for environmental inputs that may be just as important as the genetic risk in determining whether a given dog actually develops the trait being scored. A dog with a high polygenic risk score for hip dysplasia may still have healthy hips if other factors align; a dog with a low score may still develop problems if other factors do not.
Why It Matters for Your Dog
What This Cannot Predict
QTL mapping cannot turn a complex trait into a single-gene trait by force. If the biology is polygenic, no amount of sophisticated mapping will reduce it to a simple Mendelian story, and breeders who are waiting for such a reduction are waiting for something that is unlikely to arrive.
It cannot guarantee that a dog with a better genomic risk profile will remain phenotypically normal. The risk profile is a probability statement, and individual dogs at low risk can still develop the trait while individual dogs at high risk can still avoid it.
It cannot replace direct phenotypic evaluation of traits whose expression is best measured in the body, such as orthopedic structure, cardiac function, or actual behavior in adult life. Documented Phenotypic evaluation and genomic evaluation are complementary tools that work best together rather than one replacing the other.
And it cannot automatically translate across breeds. A QTL validated in one breed may not generalize to another because of different LD structures, different allele frequencies, and different genetic backgrounds.
The right use of QTL work is interpretive and probabilistic. It helps explain architecture. It may improve risk estimation when used carefully. It does not collapse complexity into certainty, and breeders who try to use it that way are overclaiming what the tool actually delivers.
Families increasingly encounter DNA claims for complex traits because the language sounds advanced and the tests are marketed aggressively. QTL mapping helps translate what those claims actually mean.
If a breeder says a dog has been tested for a complex trait, the important question is what category the test falls into. A direct causal mutation for a clear Mendelian disease is one category, and those tests can give definitive individual-level answers. A linked marker in a validated breed-specific association is a second category, and those tests can give useful probabilistic information within the breed but may fail if applied outside it. A polygenic risk estimate built from many QTL associations is a third category, and those tests give risk probabilities that are directionally meaningful but not individually diagnostic. Those three categories carry very different levels of confidence, and collapsing them under the single label of "genetic testing" is one of the most common sources of confusion in buyer conversations.
For breeders, QTL work is valuable because it confirms that meaningful progress on complex traits usually requires multiple layers of information working together. Phenotypic screening remains essential for any trait you can actually measure in the body. Line knowledge across many generations provides context that no single test can capture. Long-term record keeping turns anecdotal impressions into trend data. Population management protects diversity so that future selection has material to work with. And, where useful, genomic association data can add a layer of individual-level risk estimation on top of everything else. None of those layers alone is sufficient for complex traits, and the breeders who produce the best outcomes on polygenic targets are usually the ones who use all of them rather than treating any single tool as the complete answer.
For JB, this matters because the program's most meaningful behavioral target is not a single-locus trait. Temperament is quantitative, shaped by many genes of small effect interacting with developmental environment and individual experience. That means genomic work may eventually contribute insight into canine temperament architecture, but no honest breeder should pretend a few markers can read the whole dog, and no program should promise outcomes that the underlying biology cannot support. The program's strength lies in the combination of selection, developmental environment, and raising philosophy rather than in any claim that a genetic test could substitute for that combination.
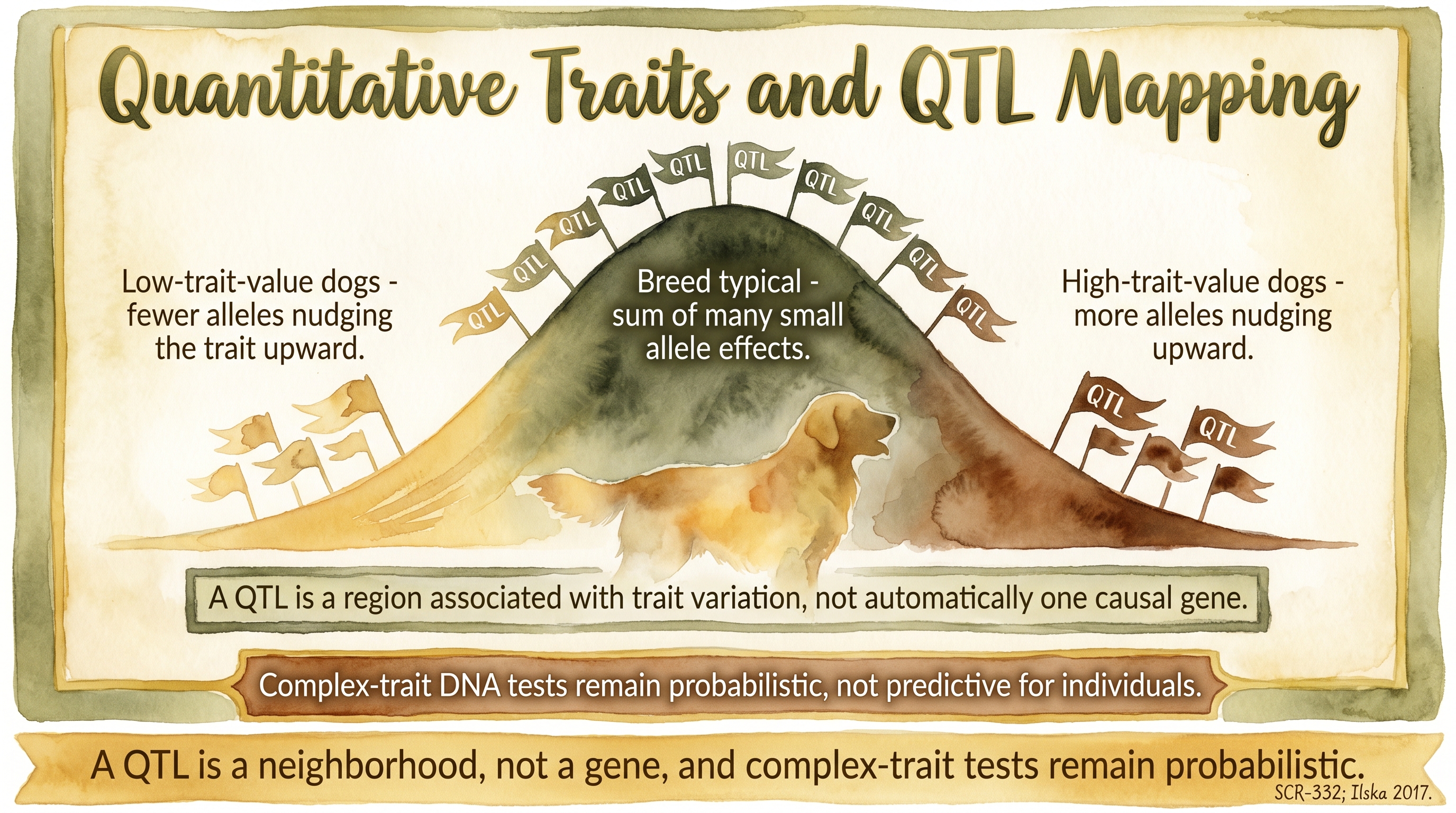
A QTL is a neighborhood, not a gene, and complex-trait tests remain probabilistic.
Key Takeaways
- Quantitative traits vary continuously and are usually shaped by many loci plus environment.
- A QTL is a genomic region associated with variation in a trait, not automatically the final causal mutation.
- Dog breeds make QTL mapping powerful because of long linkage-disequilibrium blocks, but that same structure also limits precision and cross-breed transfer.
- Complex-trait DNA tests are probability tools, not one-gene diagnoses, and should be read alongside phenotype and line knowledge.
- Polygenic risk scores can add useful information but should not be confused with definitive individual diagnoses.
The Evidence
- Quantitative-genetics frameworkgeneral genetics
Quantitative traits show continuous variation and are usually influenced by many loci plus environmental effects. - Canine genomics literaturedogs
QTL mapping and GWAS in dogs identify genomic regions associated with complex traits such as orthopedic disease, morphology, and behavior rather than simple one-gene explanations. - Canine breed-structure studiesdogs
Long linkage-disequilibrium blocks in dogs can make association mapping efficient, but they also mean associated markers are often broader regional signals rather than the final causal answer.
- Canine complex-trait literaturedogs
Polygenic risk information can inform probabilities, but it does not replace phenotypic testing or convert complex traits into binary genetic diagnoses. - Polygenic risk scoring literaturehumans and dogs
Polygenic scores are population-level probability tools whose accuracy depends on the similarity between the scoring population and the individual being scored, and they rarely generalize cleanly across breeds.
No Golden Retriever-specific study has built and validated a polygenic risk score for any common complex trait with prospective follow-up to test predictive accuracy in new individuals.
SCR References
Sources
- Ilska J., Haskell M.J., Blott S.C., Sánchez-Molano E., Polgar Z., Lofgren S.E., Clements D.N., & Wiener P. (2017). Genetic Characterization of Dog Personality Traits. Genetics, 206(2), 1101-1111. doi:10.1534/genetics.116.192674
- MacLean E.L., Snyder-Mackler N., vonHoldt B.M., & Serpell J.A. (2019). Highly heritable and functionally relevant breed differences in dog behaviour. Proceedings of the Royal Society B: Biological Sciences, 286(1912), 20190716. doi:10.1098/rspb.2019.0716
- Morrill K., Hekman J., Li X., McClure J., Logan B., Goodman L., et al. (2022). Ancestry-inclusive dog genomics challenges popular breed stereotypes. Science, 376(6592), eabk0639. doi:10.1126/science.abk0639
- Tonomura N., Elvers I., Thomas R., Megquier K., Turner-Maier J., Howald C., et al. (2015). Genome-wide Association Study Identifies Shared Risk Loci Common to Two Malignancies in Golden Retrievers. PLOS Genetics, 11(2), e1004922. doi:10.1371/journal.pgen.1004922
- Ivansson E.L., Megquier K., Kozyrev S.V., Murén E., Körberg I.B., Swofford R., et al. (2016). Variants within the SP110 nuclear body protein modify risk of canine degenerative myelopathy. Proceedings of the National Academy of Sciences, 113(22), E3091-E3100. doi:10.1073/pnas.1600084113
- Saetre P., Strandberg E., Sundgren P.-E., Pettersson U., Jazin E., & Bergström T.F. (2006). The genetic contribution to canine personality. Genes, Brain and Behavior, 5(3), 240-248. doi:10.1111/j.1601-183X.2005.00155.x